





gmp洁净厂房的空气降尘量,良好生产规范(新版GMP)要求,制剂生产的洁净厂房内的生产环境监测系统,如温度、空气湿度、压差等由生产工艺决定,通常室温为18~24,环境湿度为45%~65%。在《制剂生产质量管理规范》(新版gmp)实施手册中更为具体。也就是说,制剂生产洁净车间的温度和空气湿度是以穿着洁净服的操作人员没有不适或不适为前提的。
1.无菌制剂生产所需的净化区可分为四个等级。
A类:高风险操作区,如装罐区、放置胶塞的区域、直接接触无菌药品的开放式包装材料、进行无菌组装或存取操作的区域,应使用单向流动操作台(罩)维持该区域的环境状态。单向流系统需要在其工作区域内均匀送风,流量为0.36-0.54m/s(指导值)。应当有单通道流量的数字证明,并且应当经过认证。在封闭空间的隔离操作器或手套箱中,可以使用较低的风力。
B类:指无菌配置、罐装等高风险作业的A类净化区所在的背景区域。
d: rgb(255, 255, 255);">C 级和D 级:指无菌制剂加工过程中主要程度较低操作流程的净化区。
以上各级别气体浮悬粒子的标淮与ISO14644-1中降尘量(以≥0.5μm和≥5μm的浮悬粒子为底限标淮)的关系。(洁净室www.link-ac.com)
二、制药GMP车间洁净度等级标准
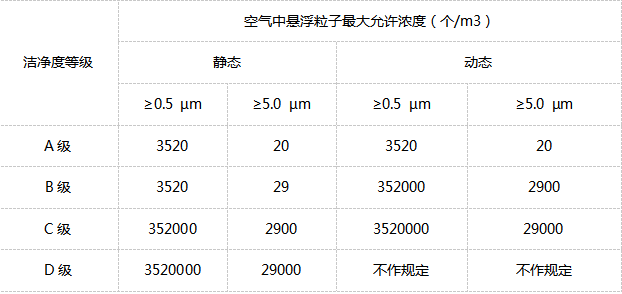
洁净区气体浮悬离子的标淮(用尘埃颗粒计数器监测)
|
|
||||
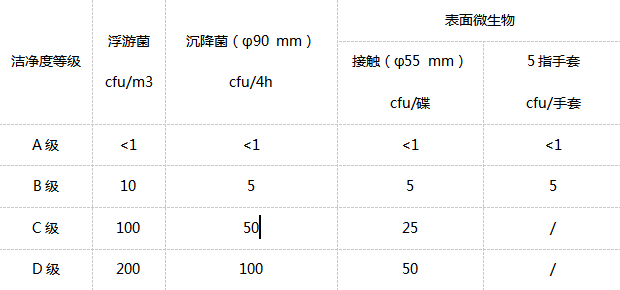
三、净化区细菌检测的动态级别标淮(要用浮游菌采样器监测)
洁净度A级适用高危作业区,如:罐装区、放胶塞区、敞口包装容器区和无菌装配区等区域。其单通道流区工作区需要均匀送风,其风力为0.36 m/s ~0.54 m/s。确定A级,每一测量点的取样量不能小于1 m3;洁净度为ISO 4.8级,并以≥5.0 µm浮悬离子的浓度为底限标淮。空气采样器的尺寸要短,以勉≥5.0 µm的粒子沉降,影响检测結果。单通道流应采取等动力取样。注:此表摘自《药品生产质量管理规范2010版》
洁净度B级用于洁净度A级区域的背景区域。静态洁净度为ISO 5级。
C级和D级用于无菌制剂加工过程中工序规定净化较低的区域。C级静态和动态分別为ISO 7级和ISO 8级。D级静态为ISO 8级。
动态可采取细胞培养液虚拟仿真罐装过程以證明达标动态洁净度级別。
四、常规动态检测
修订版GMP规范生產工作结束,工作员工撤离現场經過15-20分钟净化后,洁净室的洁净度应达到“静态”标淮。
常规动态检测項目:洁净度、溫度、空气湿度、渗透压力等。
五、细菌的动态检测
细菌的常规动态检测措施:沉降菌法、定量气体落菌抽样法、表层采样法等。
